1. 任何年龄均可发病,中位年龄40-50 岁,男性稍多。临床表现为乏力、体重减轻、盗汗、贫血、脾大;部分病例无症状,常规体检查血时发现。如不治疗,多数患者从慢性期进展为加速期再到母细胞期,也有的从慢性期突然进入母细胞期,甚至一开始就表现为母细胞期。
2. 各期共同形态学和遗传学特征:粒细胞及其前体细胞增高(母细胞计数不等),噬碱粒细胞增高,血小板高,t(9;22)(q34;q11)或其变异型染色体异位,或/和BCR-ABL1融合基因阳性(p210、p230、p190)。
3.慢性期形态学特征及诊断标准(2017年版WHO)
1)外周血:
白细胞增高,12-1000X109/L(中位值80X109/L),以中幼粒细胞和分叶核粒细胞为主;
部分病例血小板增高(p230),少数病例仅表现为血小板增高;
原始细胞<2%,嗜碱性粒细胞绝对数增多,单核细胞一般不高(<3%),但少数病例增高(p190)
2)骨髓:
细胞密度/增生程度通常显著增高(>95%);仅表现为血小板增高的病例细胞密度可正常或轻度增高;
粒系增生明显至极度活跃, 骨小梁周围不成熟细胞常增加到5-10个细胞的宽度(正常只有2-3层);
成熟谱系类似外周血,原始细胞<5%,红系减少;
巨核系可以减少,但40-50%病例巨核系可显著增多,以分叶少、单个核、体积小(“侏儒型”)为特征;
嗜酸细胞、嗜碱细胞常增多,经常见到高雪细胞;
30-40%的病例网状纤维增加伴巨核细胞增多,并与脾肿大相关。
4. 加速期:符合下列任何1项血液学/细胞遗传学标准a 或对TKI反应的临时标准即可诊断加速期(2017年版WHO):
血液学/细胞遗传学标准a
①持续或进行性白细胞增多(>10×109/L),且治疗无效;
②持续或进行性脾肿大,治疗无效;
③持续血小板增多(>1000×109/L),且治疗无效;
④持续血小板减少(<100×109/L),治疗无效;
⑤外周血嗜碱粒细胞≥20%;
⑥外周血和/或骨髓原始细胞 10%~19%b、c;
⑦初诊时Ph阳性细胞有另外克隆性染色体异常,包括所谓的主通路异常(如第二个费城染色体、8号染色体3体、17q等臂染色体,19号染色体3体),复杂核型、3q26.2异常;
⑧治疗过程中Ph阳性细胞出现任何新的克隆性染色体异常。
对TKI反应的临时标准
①血液学对第一个TKI抵抗(或未获完全缓解d)
②任何血液学、细胞遗传学或分子显示对连续2个TKI抵抗
③TKI治疗过程中发生2个或更多BCR-ABL1融合基因突变。
※a.骨髓活检中大的簇状或片状分布小巨核细胞伴显著网状纤维或胶原被认为是AP的证据,这些证据常和其他一种或多种上述列表中的改变伴随出现。
※b.外周血或骨髓中明显的淋巴母细胞(即使<10%)应警惕快速淋巴母细胞转化,需要进一步临床及遗传学检查。
※c. 外周血或骨髓原始细胞≥20%或髓外原始细胞增生浸润应诊断为CML 母细胞期。
※d.完全血液学缓解的标准:血白细胞计数<10×109/L,血小板<450×109/L, 无未成熟粒细胞,脾脏不能触及。
5. 母细胞期:母细胞期的诊断标准包括(符合其中之一即可诊断):
①外周血或骨髓原始细胞≥20%(一些学者或临床试验采用原始细胞≥30%为母细胞期的诊断标准,但不管采用哪个诊断标准,大多数母细胞期患者预后不良)。骨髓活检标本中原始细胞局部片状分布占据一定的面积,如1个完整的小梁间区或更大,即使其它区域仍表现为慢性期也要确定为母细胞期的证据;
②髓外原始细胞增生浸润。最常见于皮肤、淋巴结、骨、中枢神经系统,其它任何部位均可发生。
4. 免疫表型特征
1)大多数母细胞期病例的原始细胞为髓系,包括中性粒、单核、巨核、嗜碱粒、嗜酸粒、或者红系、或者以上任何形式的组合。
2)20-30%母细胞期患者原始细胞为淋巴母细胞(通常为B淋巴母,但也有T淋巴母和NK细胞的报道)。
3)髓系母细胞期的原始细胞MPO表达可以很强,也可以弱表达甚至不表达,但表达1种或多种粒、单核、巨核或红系分化抗原,如CD33、CD13、CD14、CD11b、CD11c、CD117、CD15、CD41、CD61、CD235a、CD235b。
4)很多髓系母细胞期病例,原始细胞也表达1种或多种淋巴细胞相关抗原。
5)多数淋巴母细胞转化病例为B淋巴母源性,除表达CD19、CD10、CD79a、PAX5、CD20等B细胞相关抗原外,还表达TDT。少数病例为T淋巴母源性,表达CD2、CD3、CD5、CD7、CD4、CD8等T细胞相关抗原。
6)淋巴母细胞期病例常表达1种或多种髓系相关抗原,也可见双系来源(明显的髓系细胞群和淋系细胞群),也有序贯发生B淋巴母和髓系母细胞转化的病例报道。
7)近期研究显示TKI治疗后发生少见母细胞类型转化的病例不断增加,如原始嗜碱粒细胞、原始巨核细胞等。
8)慢性期CD34阳性细胞表达CD7为预后不良因素,而CD34阳性细胞群比例正常、不表达CD56、CD7或CD11b等异常标记预示对TKI治疗敏感。
5. 遗传学
1)90-95%病例都有特征性的t(9;22)(q34.1;q11.2),其余病例表现为涉及第3个甚至第4个染色体变异型异位或其变异型染色体异位,或9q34.1与22q11.2隐性异位(常规染色体检查不能发现,需要FISH或RT-PCR检测)。
2)根据BCR基因的不同断裂点部位,产生三种大小不同的融合基因蛋白:p210、p190、p230,p230型常显示中性粒细胞明显分化成熟和/或血小板增多,而p190型常与单核细胞增多相关。
3)80%病例在AP或BP转化时显示除ph染色体外的染色体异常,包括另外Ph染色体、+8、+19、i(17q)等,CML初诊时有上述任何染色体异常提示预后不良,故将这些病例划分为AP期。
4)AP及BP转化过程中常涉及基因突变,包括TP53、RB1、MYC、CDKN2A(p16INK4a)、NRAS、KRAS、RUNX1(AML1)、TET2、CBL、ASXL1、IDH1和IDH2,但这些基因突变在转化过程中的作用还有待进一步研究。
6. 预后
未接受有效治疗的病人大多数于3-5年转化为AP、BP。TKI治疗CML预后的最重要影响因素是在血液学、细胞遗传学和分子水平对TKI治疗的抵抗。第一代TKI治疗遗传学完全反应率为70-90%,5年无进展和总生存率为80-95%。二代TKI治疗可更快更高比例获得分子水平缓解。TKI治疗时代很少CML患者死于白血病,总体生存率和无白血病人群近似。
43岁男性外周血白细胞显著增高

53男性,CML-chronic phase




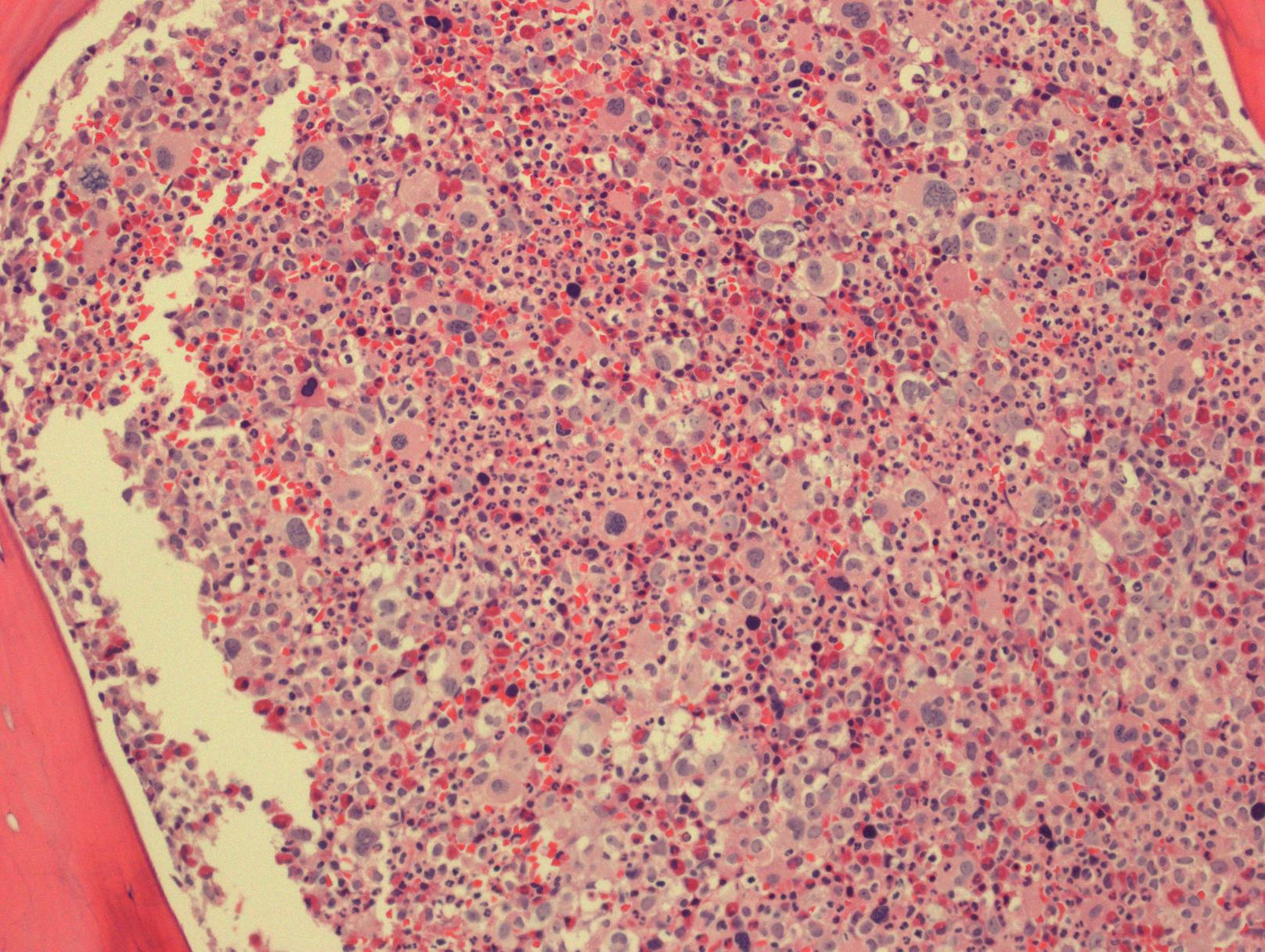
73女性,CML with blastic transformation

2.不典型慢性粒细胞白血病,BCR-ABL阴性:外周血白细胞持续性增高(至少13 x 10⁹/L),其中不成熟粒细胞(早幼粒、中幼粒、晚幼粒)至少10%,并有发育异常;骨髓粒系增生伴发育异常。部分病例有SETBP1突变,无t(9;22) (q34;q11) 或其变异型染色体异位,BCR-ABL1融合基因阴性。
3.慢性粒-单核细胞白血病:外周血持续性单核细胞增高至少达1 x 10⁹/L,并同时有外周血或骨髓一系或多系发育异常,或有克隆性细胞/分子遗传异常,但无t(9;22) (q34;q11) 或其变异型染色体异位,BCR-ABL1融合基因阴性。
4.反应性粒细胞增高/类白血病反应: 有相应病史,如感染。外周血粒系增高,可有核左移,常有中毒性改变,噬碱粒细胞不高,中粒细胞碱性磷酸酶活性正常。无t(9;22) (q34;q11) 或其变异型染色体异位,BCR-ABL1融合基因阴性。