几乎全部阳性(≥95%的病例阳性):
Fascin、FVIIIA、FXIIIA、CD163、CD45、CD68、Vimentin通常阳性(<95%,≥75%的病例阳性):
Cathepsin-K、CD4经常阳性(<75%,≥55%的病例阳性):
CD63、lysozyme有时阳性(<55%,≥35%的病例阳性):
CD31少数阳性(<35%,≥15%的病例阳性):
CD15偶尔阳性(<15%,≥5%的病例阳性):
actin-HHF-35、S100几乎全部阴性(<5%的病例阳性):
FVIIIRAg2. 无论发生在儿童或成人,病变的组织学基本相似。病变位于真皮内,可延伸至表皮,但不侵犯表皮层,也可向皮下的脂肪组织甚至横纹肌组织浸润;位于深部的病变相对界限较清楚;
3. 镜下可见成片、致密的单核样组织细胞增生,常紧紧包绕汗腺、皮脂腺、毛囊等附属器;
4. 疾病的不同阶段病理改变有所不同,据此可分为早期JXG、经典型JXG、移行JXG和混合型JXG四种类型:
5. 早期JXG
1)主要由片状增生的单核样组织细胞组成,胞质少至中等量,呈均质双色或淡嗜伊红色,胞质内脂质空泡不明显;
2)核小,圆形和卵圆形,部分瘤细胞可见核沟,多数核含有一个小的核仁;
3)无明显核多形性或异型性,可见数量不等的核分裂像,但无病理性核分裂像;
4)少或无杜顿巨细胞;
6. 经典型JXG
1) 单核样组织细胞胞质丰富,可见脂质空泡,或呈黄色瘤细胞样;
2) 核呈不规则形或肾形,核膜清晰、可见核沟,染色质空可见核仁,核分裂象罕见;
3) 常可见多少不等的经典型杜顿巨细胞、早期的杜顿巨细胞以及朗罕型多核巨细胞;
7. 移行JXG
1)多出现在内脏或疾病的晚期;
2)主要由梭形细胞组成,也称梭形细胞(纤维组织细胞性)黄色肉芽肿;
3)梭形细胞常呈席纹状排列,伴有泡沫样组织细胞和多核巨细胞;
4)间质可伴有纤维化;
8. 混合型JXG:同一病例包括上述两种或三种不同亚型形态学特征;
9. 间质内可见一定数量的急、慢性炎症细胞浸润,特别是嗜酸性细胞,易与朗格汉斯细胞组织细胞增生症相混淆;
10. 被覆的表皮可变扁平;
11. 发生皮肤以外部位的病例杜顿巨细胞的数量较少或缺如。
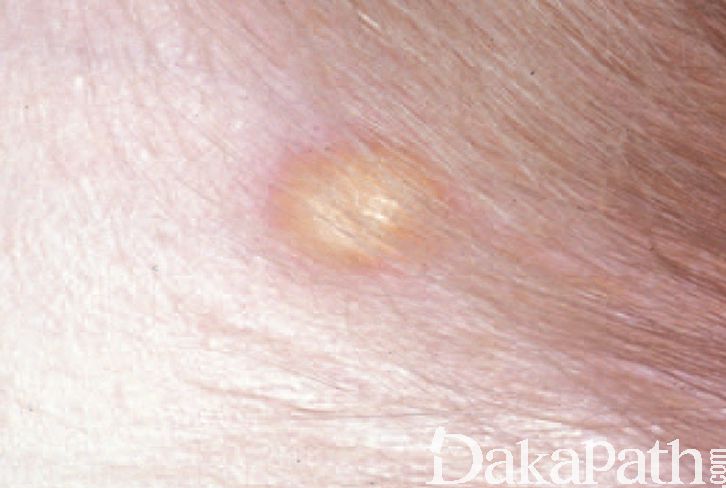
位于头皮的孤立性黄色结节
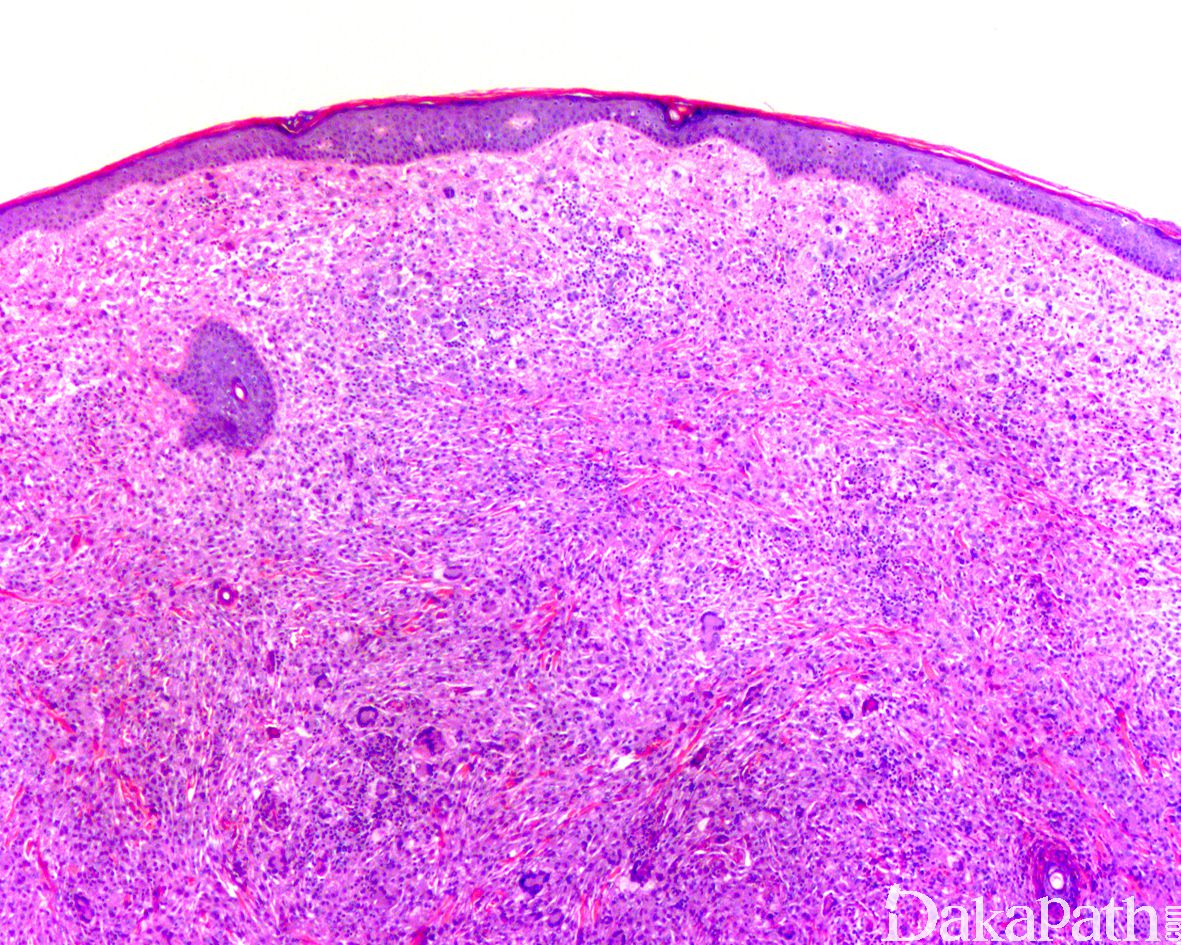
病变比邻表皮下生长,围绕皮肤附属器

早期非脂化的JXG,见成片的单核样组织细胞增生,偶见早期的杜顿型巨细胞

成熟的JXG,可见成片的杜顿型巨细胞
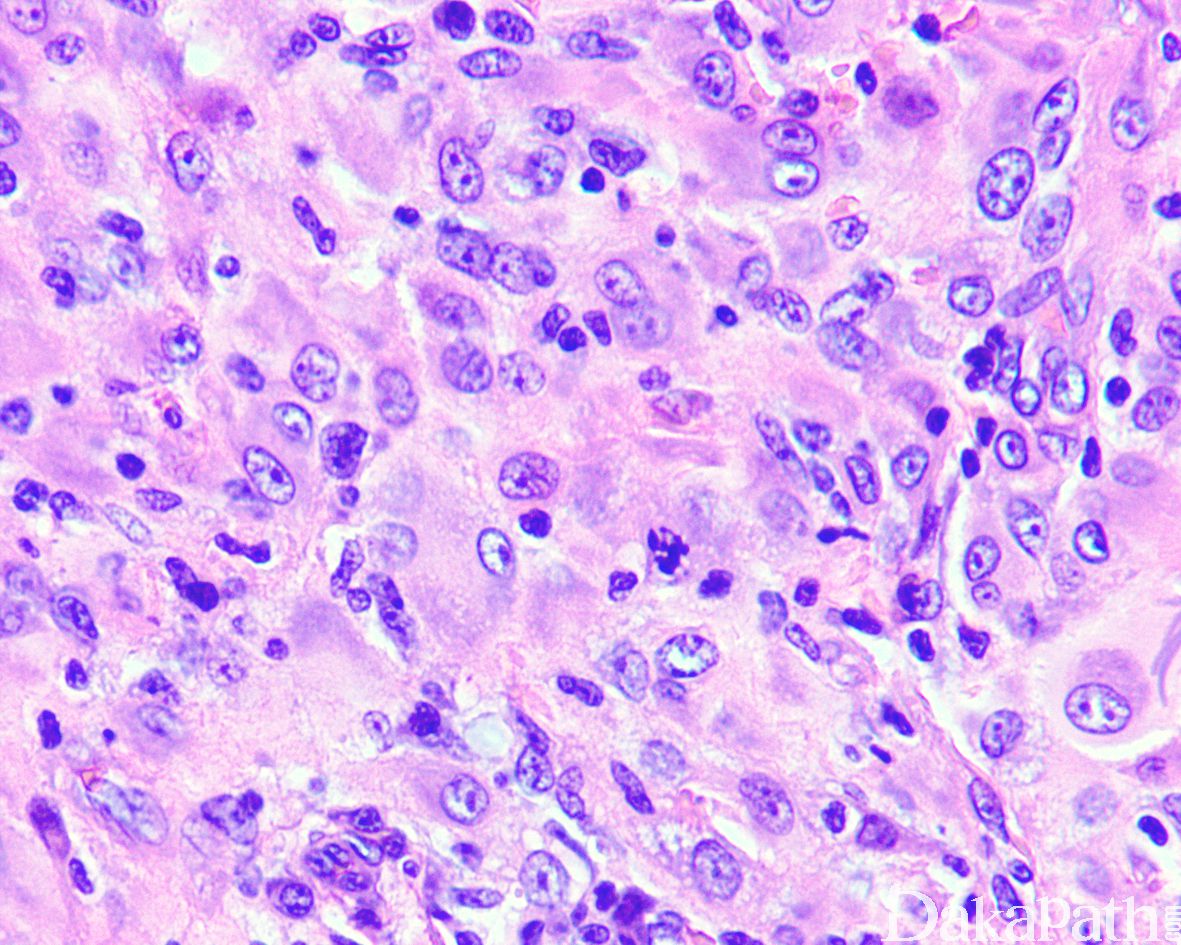
偶尔可见核分裂象
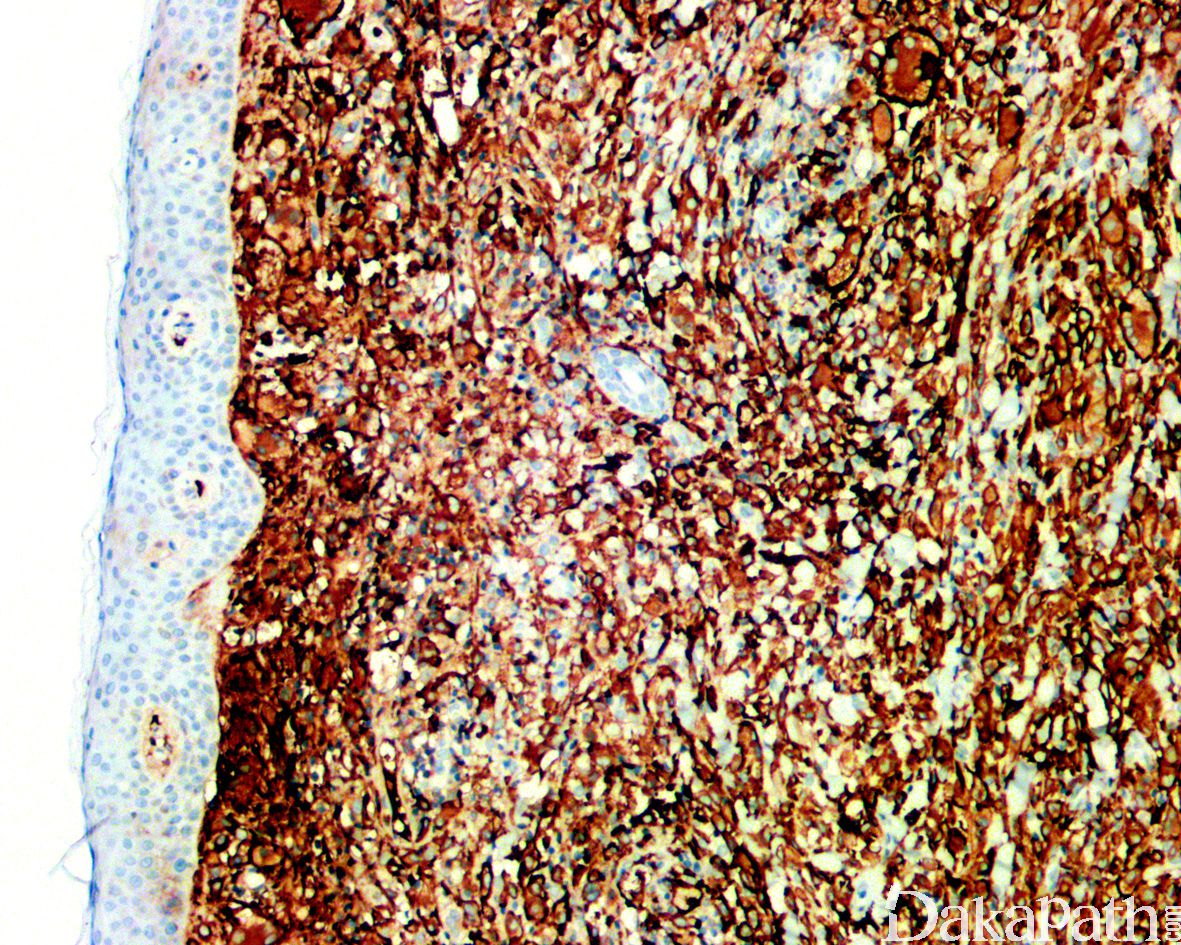
免疫组化染色弥漫表达CD163

单核样组织细胞增生伴有数量不等的嗜酸性粒细胞浸润

表达CD14
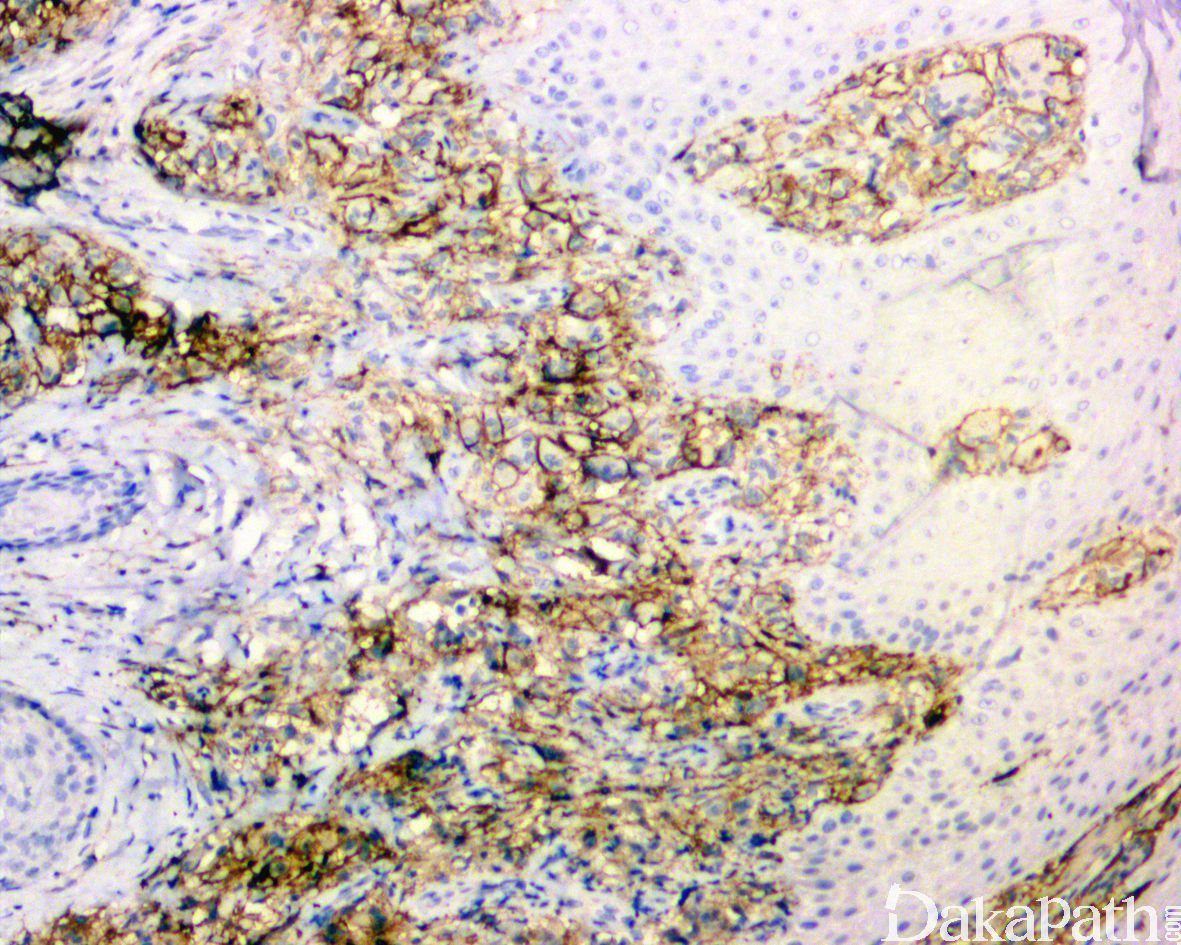
表达CD4

SMA阴性
1.皮肤朗格汉斯细胞组织细胞增生症(LCH):早期JXG和LCH的鉴别诊断诊断困难,JXG不浸润表皮,构成细胞黏附性强,少见嗜酸性粒细胞浸润,可见杜顿型巨细胞;皮肤LCH常见累及表皮,构成细胞黏附性差,嗜酸性粒细胞浸润较多,无杜顿型巨细胞;免疫组织化学:JXG FXIIIa阳性,S100和CD1a阴性,与LCH相反。
2. 黄色瘤:均匀一致的黄瘤样泡沫细胞,无杜顿型巨细胞和急慢性炎细胞浸润。
2.纤维组织细胞瘤:好发于成年人,以增生的梭形纤维母细胞为主,呈条束状或席纹状胞排列,无典型朗格汉斯细胞核特征的细胞,与LCH相反S100和CD1a阴性。
[1]Sandell R F, Carter J M, Folpe A L. Solitary (juvenile) xanthogranuloma: a comprehensive immunohistochemical study emphasizing recently developed markers of histiocytic lineage [J]. Human Pathology, 2015, 46(9):1390-1397.
[2]Janssen D, Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the kiel pediatric tumor registry. Am J Surg Pathol 2005;29:21-8.
[3]Dehner L P. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations[J]. American Journal of Surgical Pathology, 2003, 27(5):579-593.