皮肤朗格汉斯细胞组织细胞增生症
Langerhans Cell Histiocytosis
于励民
发布时间:2016-03-13 05:35:25
概述:
发病部位:皮肤、骨髓及骨组织、中枢神经系统,其他器官包括肺脏、淋巴及肝脏、胸腺及胃肠道。
诊断要点:
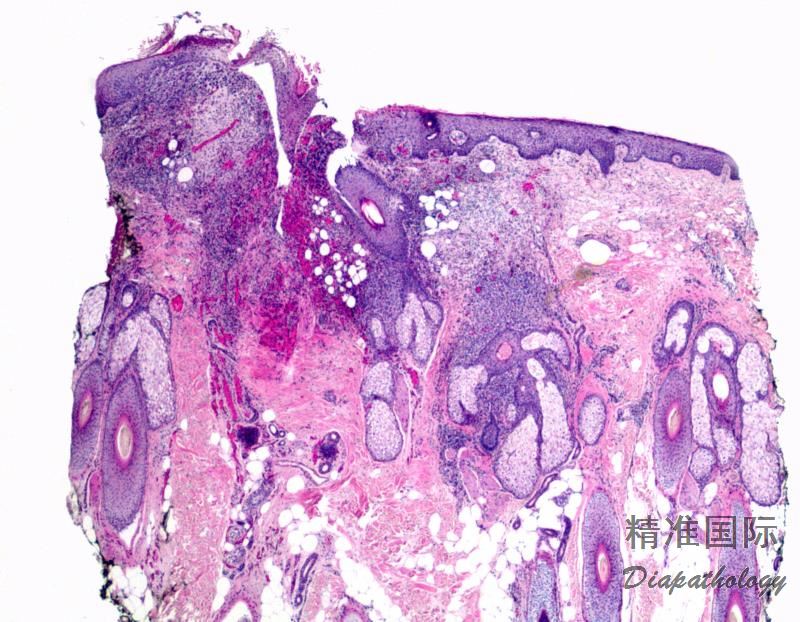
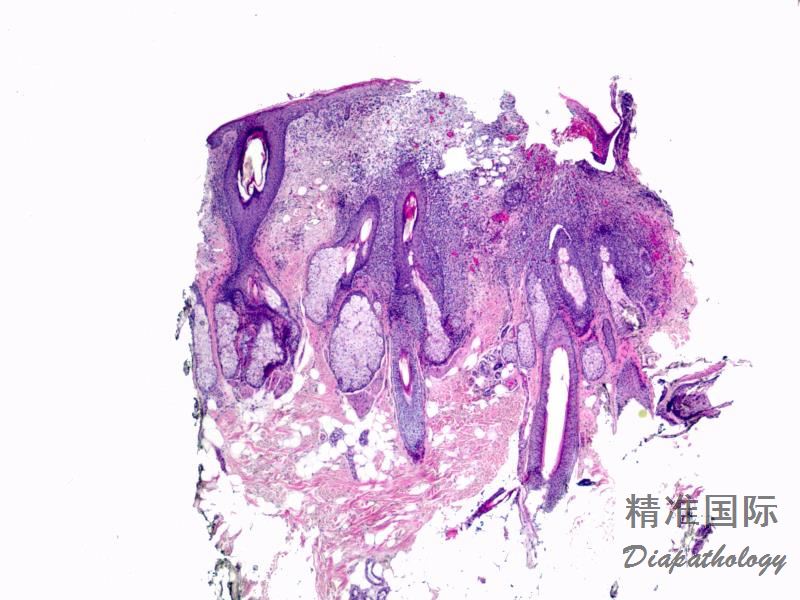
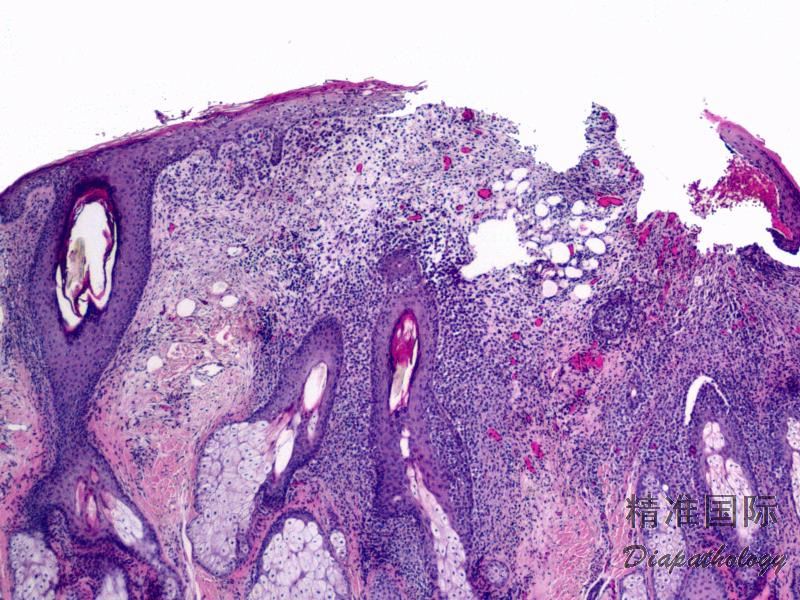
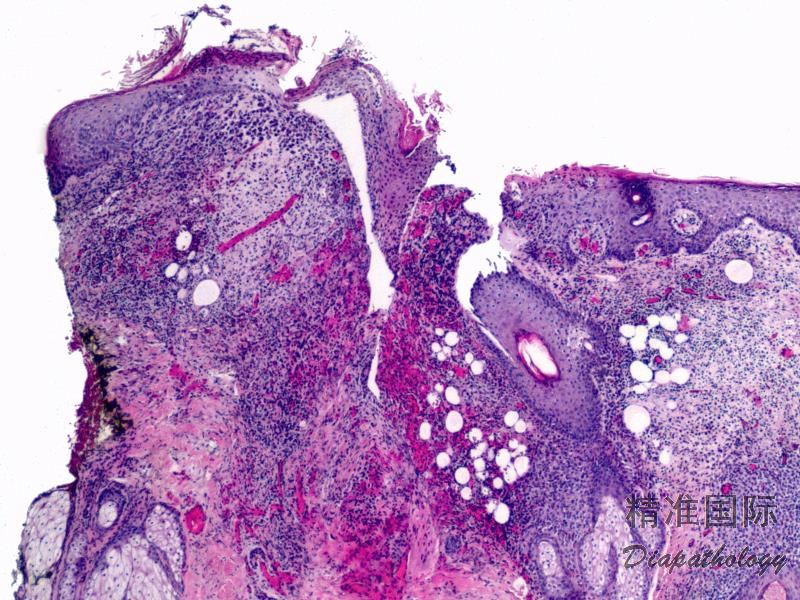
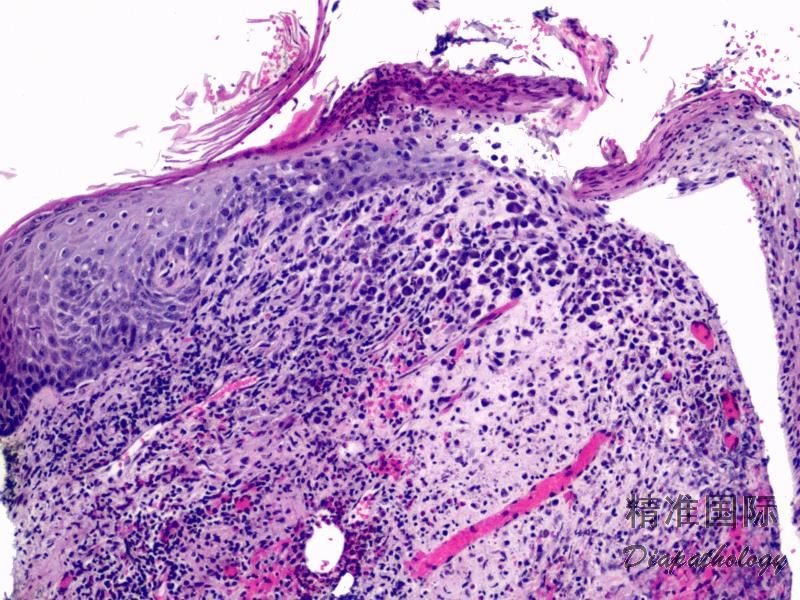
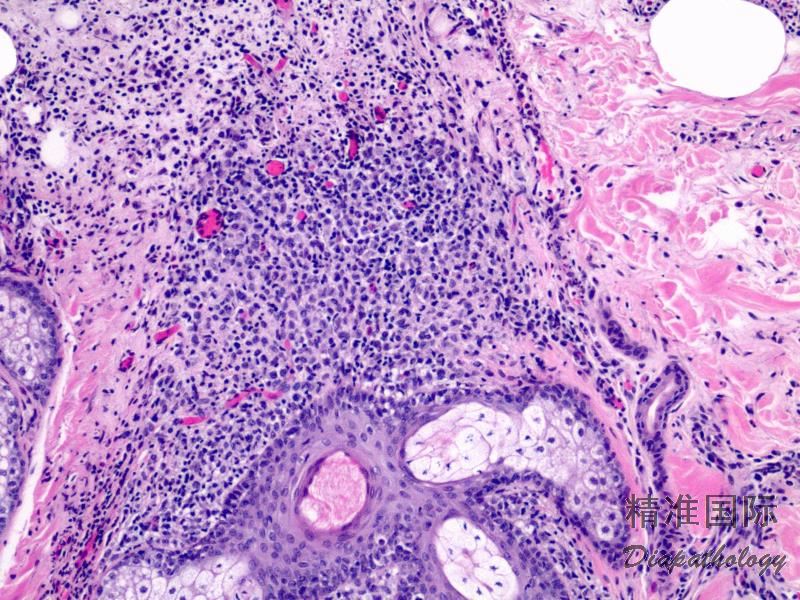
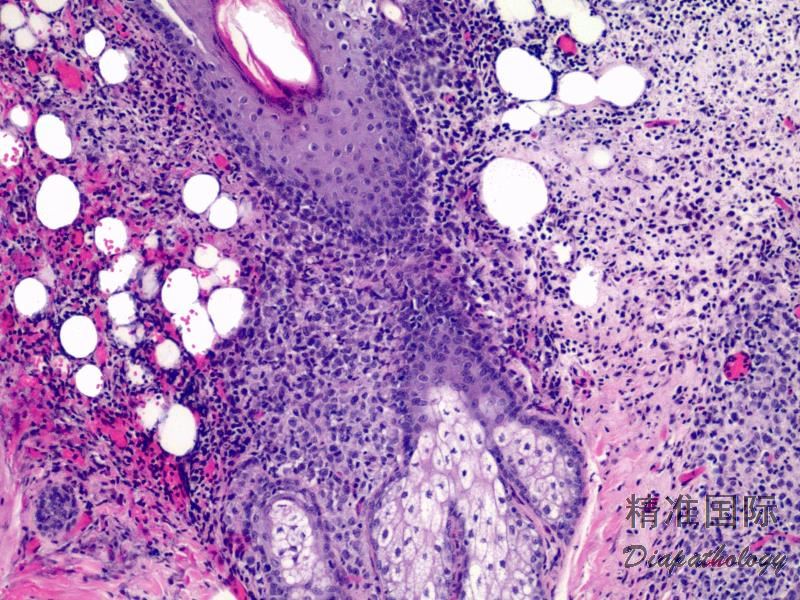
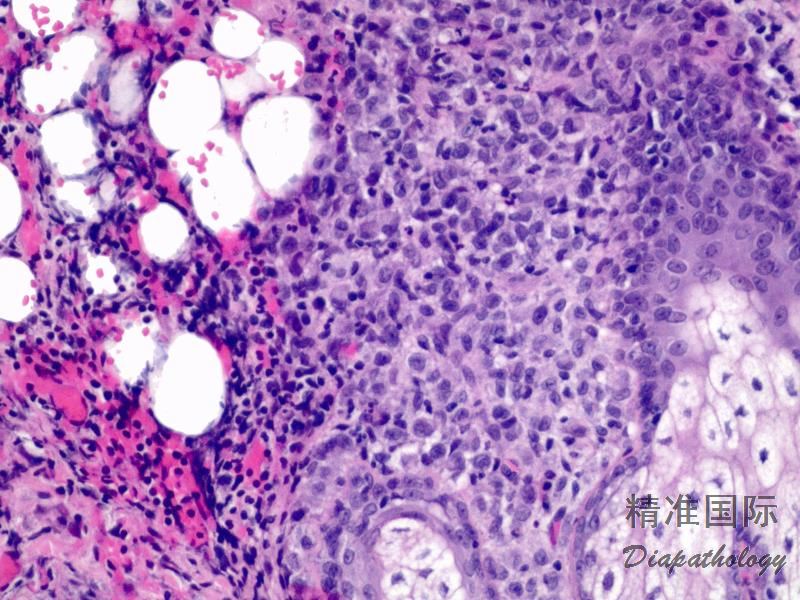
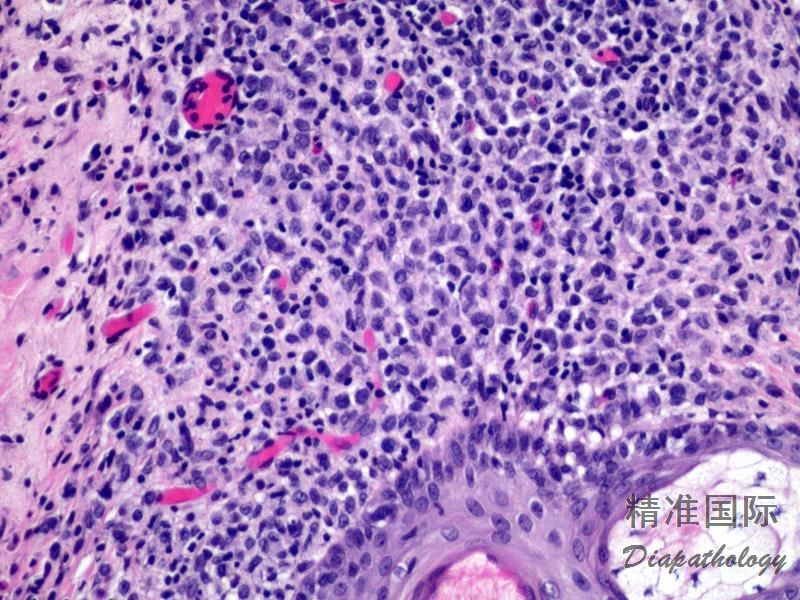
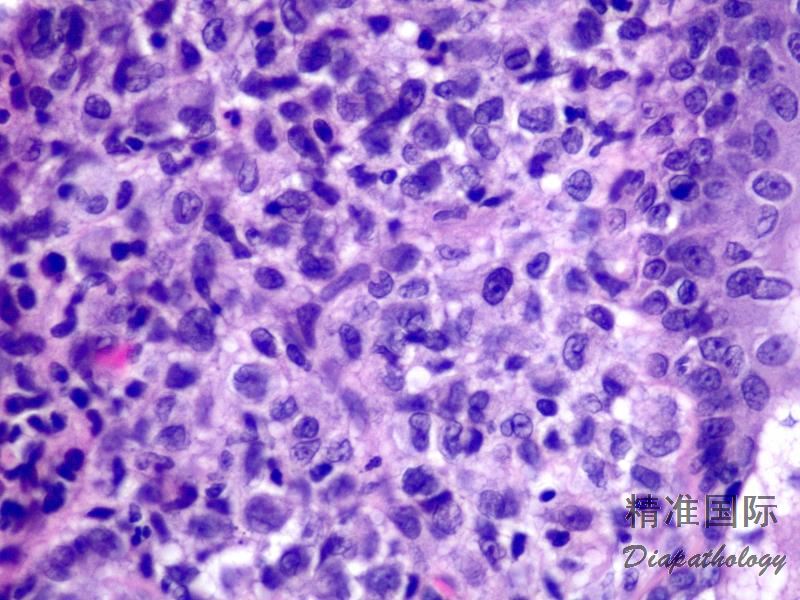
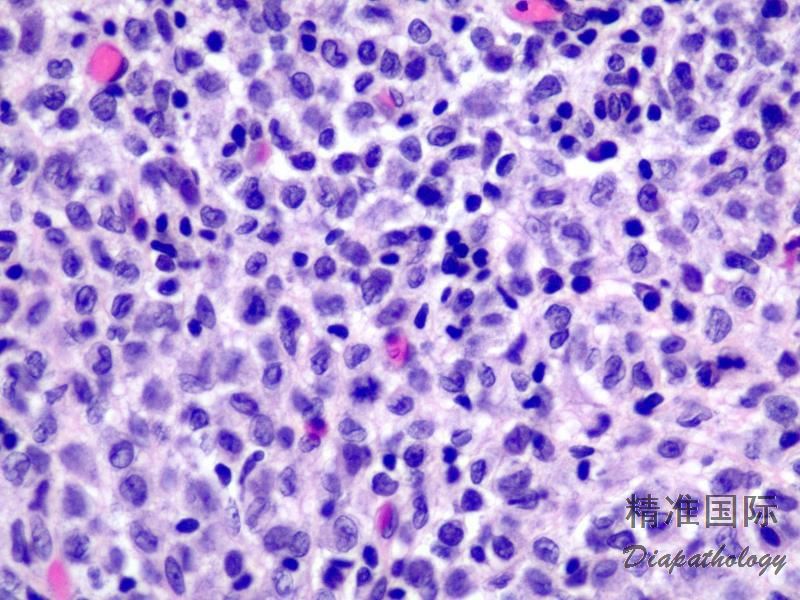
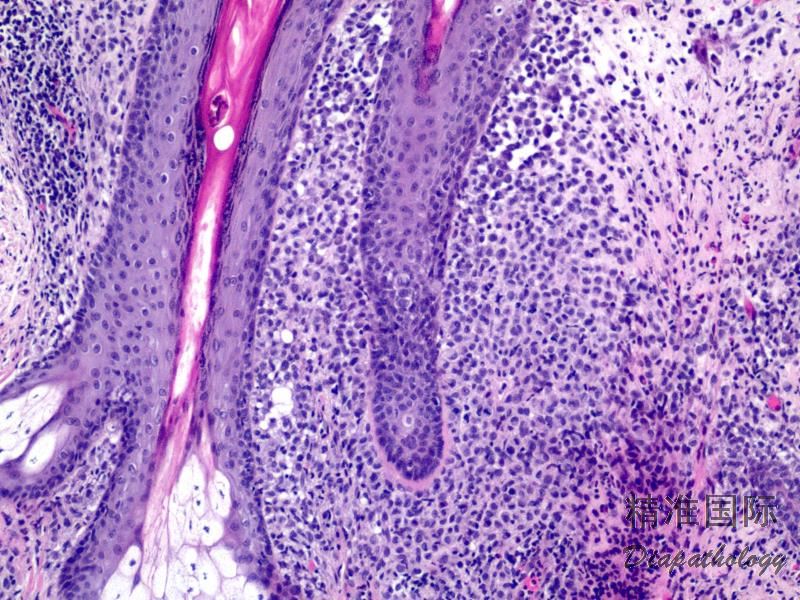
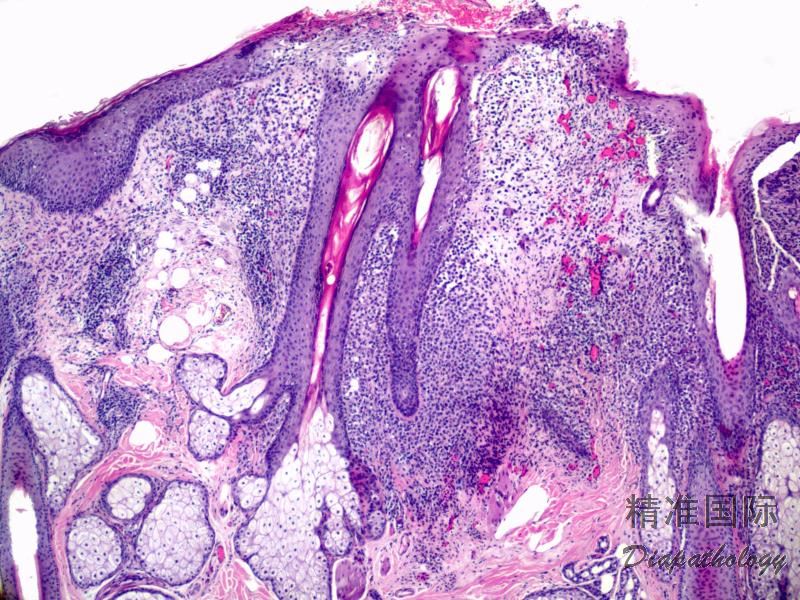
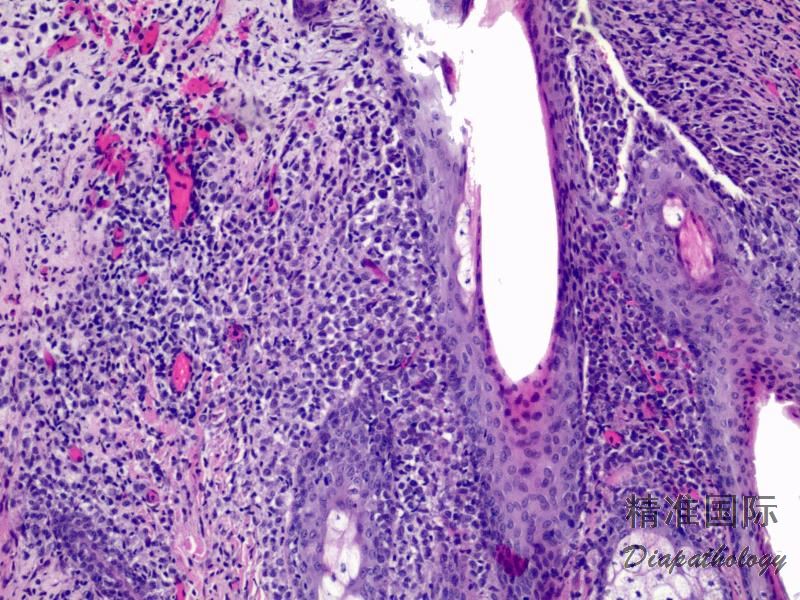
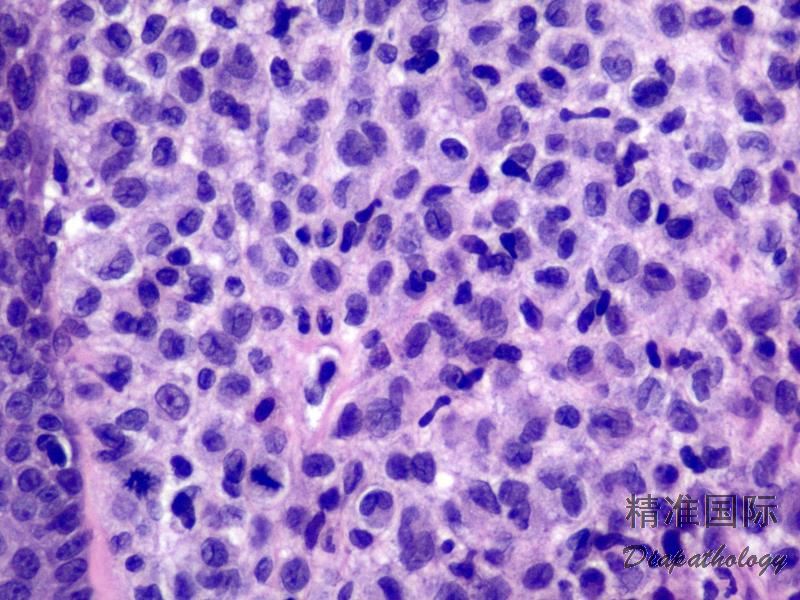
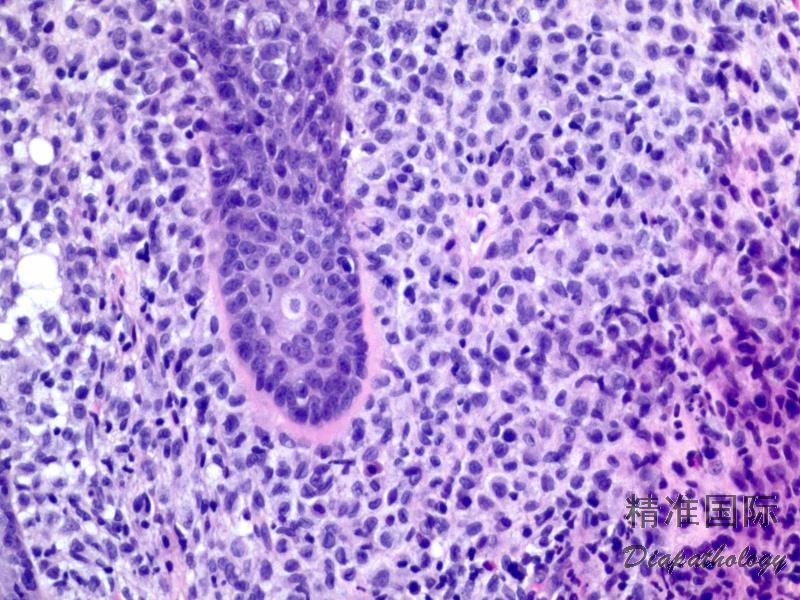
2.组织学上真皮内及皮下朗格汉细胞浸润,常延伸至表皮。朗格汉细胞显示丰富嗜酸性胞浆,伴核沟形成的咖啡豆样或叶状胞核,可见小核仁。常见大量嗜酸性粒细胞浸润,亦可见其他炎细胞浸润(包括多核巨细胞)。常见坏死及核分裂(常无不典型核分裂)。淋巴结结构尚存,窦内可见朗格汉细胞浸润。骨髓中可显示单个或小簇状朗格汉细胞与造血前体细胞混合性分布。肝脏表现为汇管区或肝实质结节,或窦内弥漫性浸润,可导致硬化性胆管炎(约15%的儿童硬化性胆管炎由LCH引起)。陈旧性病变可见坏死及纤维化增多。
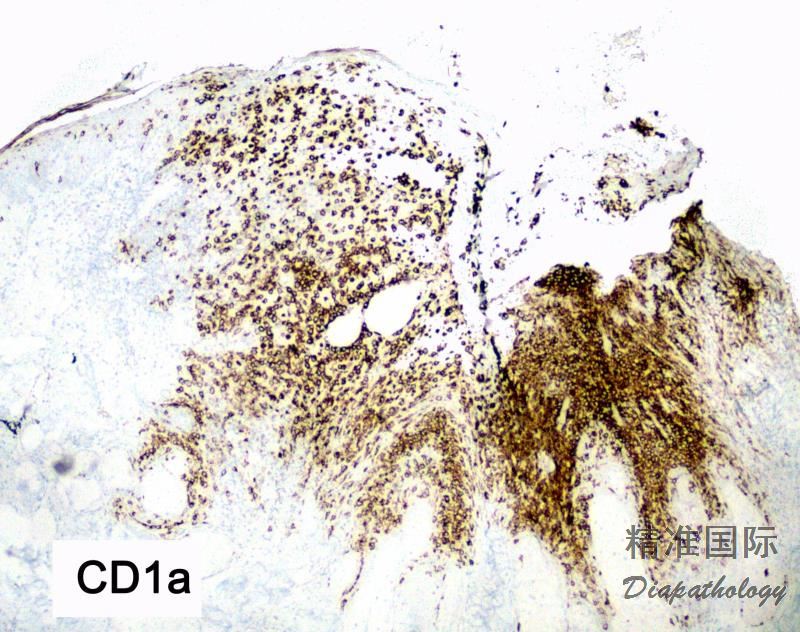
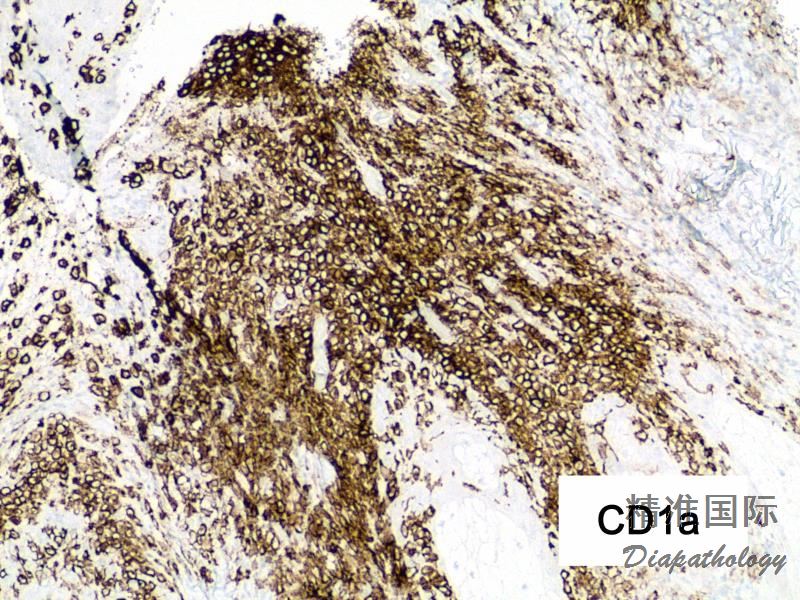
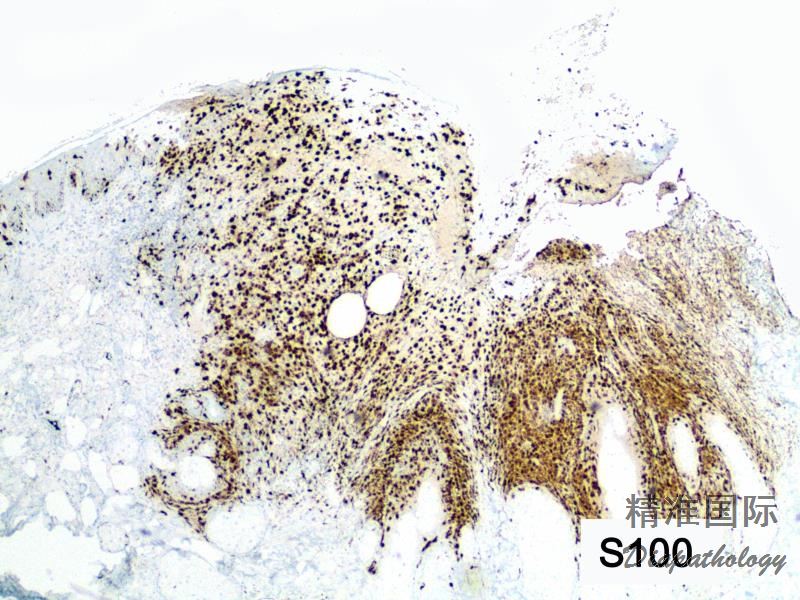
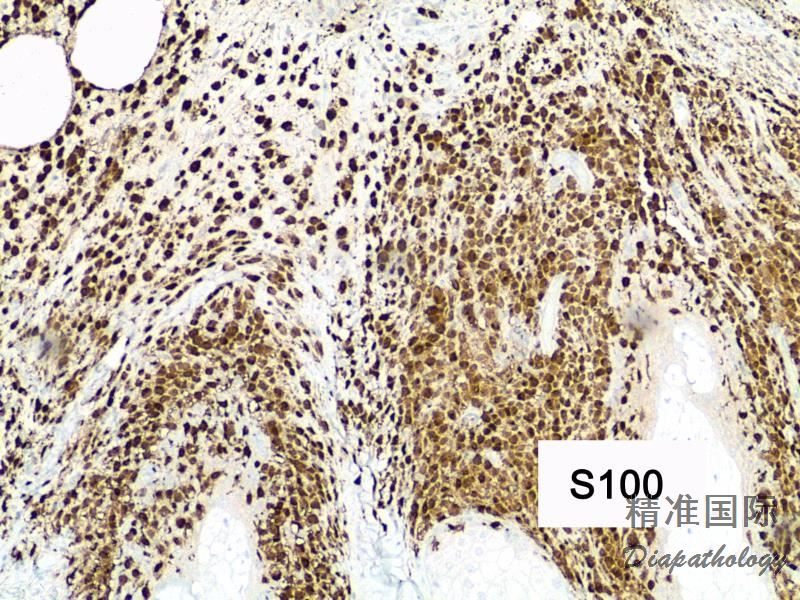
免疫组织化学染色:
鉴别诊断:
2.Rosai-Dorfman病:LCH常无核吞噬现象,组织细胞显示CD68阳性,而CD1a阴性。
3.不能明确细胞组织细胞增生症(Indeterminate Cell Histiocytosis) :组织学及免疫特点类似于LCH,超微镜下缺乏Birbeck颗粒。
4.霍奇金及非霍奇金淋巴瘤细胞:缺乏LCH细胞核特点,肿瘤细胞常呈CD15及CD30阳性,而CD1a阴性。