真性红细胞增多症
Polycythemia Vera (PV)
夏成青,陈红梅,查宏斌
发布时间:2017-03-25 03:01:13
概述:
诊断要点:
1. 临床表现和形态学
1)发病率地区差异大,全球平均发病率每100000人群年发病0.84例,男女比例1-2:1,诊断时中位年龄60岁。
2)最常见症状与红细胞容量增加所导致的高血压和血管异常有关如头痛、视觉障碍、血栓形成、出血等,肠系膜、门静脉或脾静脉血栓或Budd-Chiari综合征患者要考虑PV为潜在原因的可能。
3)外周血:正细胞、正色素红细胞轻度-明显增多,如存在缺铁则表现为小细胞低色素红细胞,中性粒细胞增多,可罕见嗜碱粒细胞增高,偶尔可见不成熟粒细胞(中、晚、杆状核),但通常不见原始细胞。
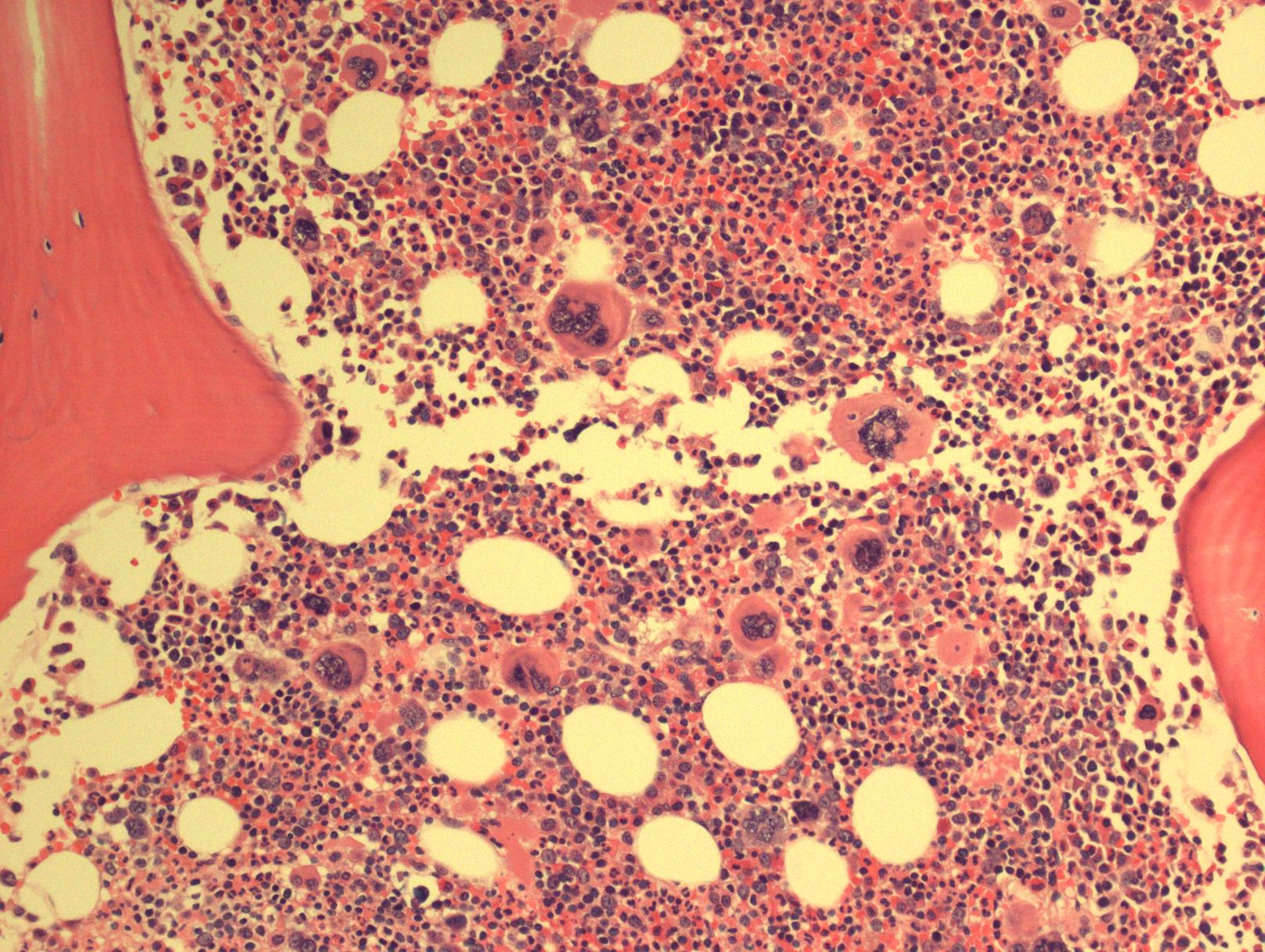
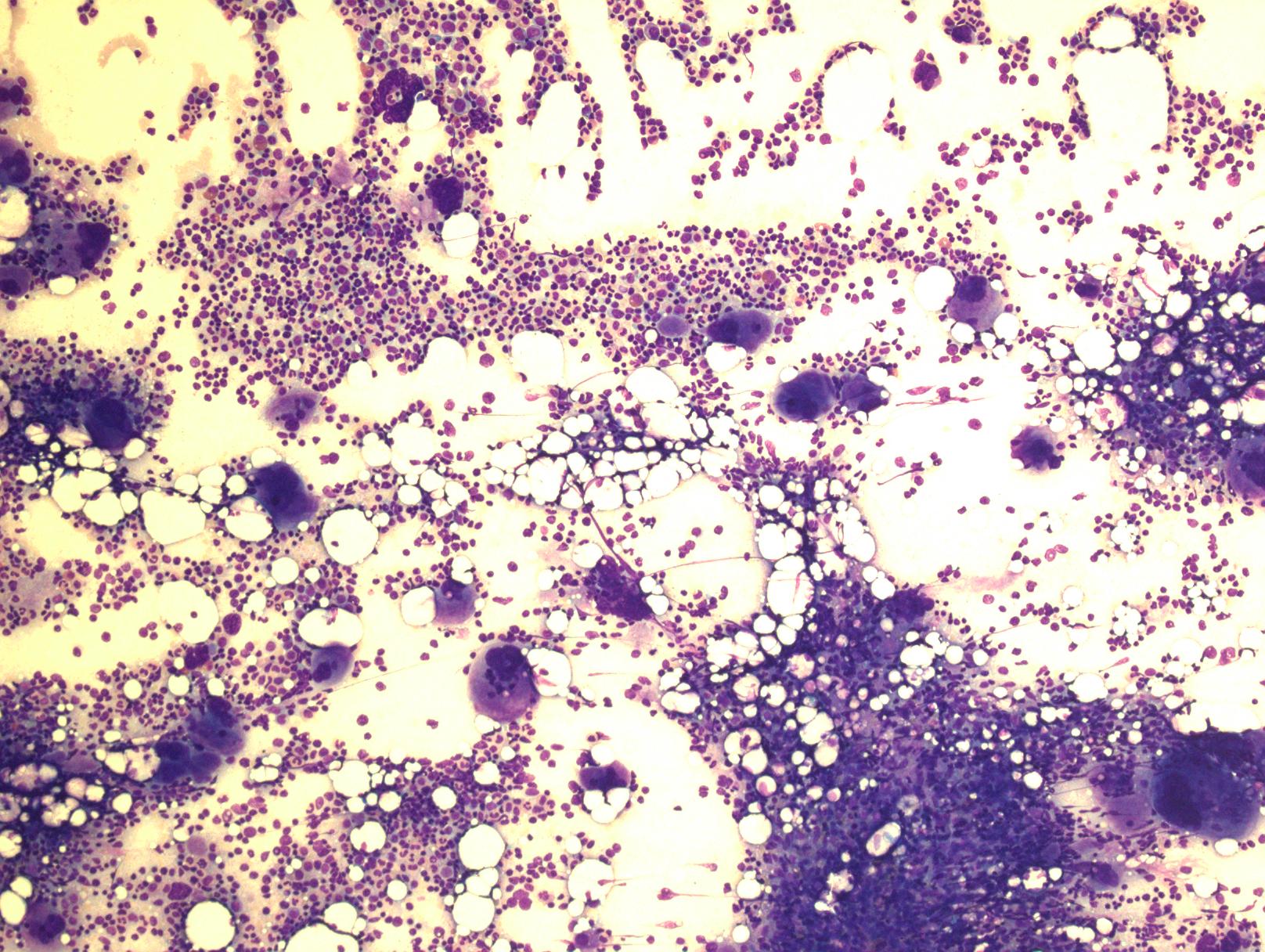
4)骨髓细胞密度增加,特别是皮质旁细胞密度增加应值得重视。通常表现为全髓系增生,但以红系及巨核细胞增生为突出。特征性表现为:红系增生为正常红细胞,红系前体细胞呈大的岛状或片状分布。粒系增生形态正常,原始细胞不增多。巨核细胞数量增加,特别是血小板增多病例,常表现为多叶核。血小板增多、血红蛋白或红细胞压积较低病例早期骨髓表现和原发性血小板增多症相似。巨核细胞常形成松散的簇状或邻近骨小梁分布,细胞呈多形性,大小不一致,大多数巨核细胞核折叠正常或分叶深,缺乏明显异型,仅少数细胞核圆形或其它异常形态,特别是伴轻微网状纤维增多时。血窦扩张,常充满红细胞。特征性的骨髓组织学改变常可将PV区别于原发性血小板增多、原发性骨髓纤维化及反应性红细胞增多或血小板增多。80%病例网状纤维网正常,其余病例可有网状纤维增多,甚至轻-中度胶原纤维增生。初诊时即使轻度的网状纤维增多(I级)也预示疾病可快速进展到PV后骨髓纤维化,因此骨髓活检很重要。超过90%病例骨髓铁染色阴性。
5)耗竭期及PV后骨髓纤维化:PV晚期红系增生趋于逐步转向减低,血红蛋白也经历正常到贫血,脾脏进一步增大。PV后骨髓纤维化转化前或前后白细胞增高预示疾病更具侵袭性。这种变化常伴随着骨髓的改变。疾病进展最常见的表现是PV后骨髓纤维化,伴随的特征性改变是外周血白细胞增多,出现不成熟粒细胞和不成熟(有核)红细胞,可见异常红细胞如泪滴样红细胞及脾脏肿大。标志性改变是明显的网状纤维和胶原纤维化。细胞密度不等,常表现为低增生。可见簇状巨核细胞,常见明显核异型、深染。有时扩张的髓窦内见红细胞、白细胞、巨核细胞聚集。不成熟细胞增多,但如果外周血或骨髓原始细胞≥10%,或者出现明显骨髓增生异常,均为不寻常表现,最大可能是已转化为加速期,原始细胞≥20%考虑为PV后骨髓纤维化母细胞期(以前称为急性白血病)。
2. 诊断标准(2017年版WHO)
多血期诊断标准
主要标准
1)血红蛋白浓度增高(男>165g/L, 女>160g/L)或 红细胞压积增高(男>49%,女>48%),红细胞压积高于正常预期值25%以上;
2)骨髓活检细胞密度较同年龄组增高,三系增生(全髓系增生),包括显著的红系、粒系、巨核细胞增生,巨核细胞多形性及分化成熟(大小不一);
3)JAK2 V617F或JAK2 第12外显子突变。
次要标准:血清促红细胞生成素低于正常水平。
满足上述3个主要标准,或前两个主要标准加上次要标准可明确诊断※
※ 如果满足主要标准“3”和次要标准,且持续红细胞绝对增高(男性血红蛋白>185g/L,女性血红蛋白>165g/L,或红细胞压积男>55.5%,女>49.5%)时,主要标准“2”(骨髓活检)可以不做。但是初期骨髓纤维化(见于20%的患者)只有通过骨髓活检才能发现,骨髓纤维化预示快速进展到明显骨髓纤维化(PV后骨髓纤维化)。
PV后骨髓纤维化诊断标准
必须标准
1)先前有WHO定义的PV诊断证据
2)骨髓纤维化(3级分类的2-3级或4级分类的3-4级)
附加标准(满足2条)
1)贫血(低于相应年龄、性别及海拔的正常范围)或者持续不需要放血(未进行减细胞治疗)或持续不需要减少细胞治疗;
2)外周血白细胞增多,出现不成熟粒白细胞和红细胞;
3)脾肿大:脾脏触诊肋下>5cm,或新出现可触及的脾脏增大;
4)出现以下症状的任意2个或全部3个:6个月内体重减少>10%、盗汗、原因不明发热(>37.5℃)
3. 细胞遗传学
95%以上病例有JAK2 V617突变,另有3%左右的病例JAK2 12外显子突变。罕见JAK2 突变PV病例获得BCR-ABL1重排,其临床意义不详,但存在形态学和血液学向CML转变的情况。诊断时约20%的病例有细胞遗传学异常,最常见的是+8、+9、-20q、-13q、-9p,随着疾病的进展,细胞遗传异常逐步增多,PV后骨髓纤维化期80-90%病例具有细胞遗传学异常,包括治疗相关MDS及急性髓系白血病常见的细胞遗传学异常。
鉴别诊断:
1.真性原发红细胞增多症(“True” Primary Polycythemia),先天性: 真性原发红细胞增多症分为先天性和获得性。本节所讨论的“真性红细胞增多症” (Polycythemia Vera)是获得性真性原发性红细胞增生症。先天性真性原发红细胞增多症患者有遗传和家族史, 系由于EPOR基因突变所致,红细胞增多,但无粒系和巨核系增生。
2.真性继发性红细胞增多症(“Ture” Secondary Polycythemia):也包括先天性和获得性,前者如2,3-二酸变位酶(2,3-BPG)缺乏,后者多见于缺氧如慢性阻塞性肺疾病以及有的肿瘤产生的EPO。
3.假性红细胞增多症(“False”Polycythemia):由与脱水等原因造成的血液浓缩所致,红细胞容量(压积)不高。
4.原发性血小板增多症:需与早期PV相鉴别。原发性血小板增生症主要是血小板增高,红细胞或血红蛋白不增高。骨髓细胞密度正常,仅见巨核细胞增生且异型性不明显,粒、红系无增生。
5.原发性骨髓纤维化:较早期原发性骨髓纤维化骨髓巨核细胞增生伴显著异形,但红细胞或血红蛋白不增高;晚期原发性骨髓纤维化与晚期PV形态学常难以区分,但病史有助鉴别。
预后: